


Thẩm định trong Sản xuất Dược phẩm
Thẩm định được định nghĩa như sau:
“Một chương trình được ghi chép văn bản cung cấp đảm bảo mức độ cao rằng một quy trình, phương pháp hoặc hệ thống cụ thể sẽ tạo ra kết quả đáp ứng các tiêu chí chấp nhận được xác định trước một cách nhất quán. (ICH Q7A)”
“Thiết lập bằng chứng được ghi chép cung cấp đảm bảo mức độ cao rằng một quy trình cụ thể sẽ luôn tạo ra sản phẩm đáp ứng các thông số kỹ thuật và thuộc tính chất lượng được xác định trước một cách nhất quán. (Hướng dẫn Thẩm định quy trình của FDA năm 1987)”
Thực hiện Thẩm định hiệu quả và chất lượng cao là một trong những thách thức quan trọng nhất cần đạt được trong các dự án xây dựng mới hoặc cải tạo các nhà máy dược phẩm. Gần đây, ngành công nghiệp dược phẩm đã chuyển đổi từ Thẩm định truyền thống sang “Thẩm định dựa trên khoa học và rủi ro”. Quá trình chuyển đổi này đòi hỏi phải triển khai đánh giá rủi ro và tạo ra các kế hoạch Thẩm định dựa trên các đánh giá này, tăng cường nỗ lực cần thiết cho quá trình Thẩm định.
CM Plus cung cấp các dịch vụ phù hợp với nhu cầu của khách hàng, từ hỗ trợ tạo kế hoạch Thẩm định gốc và đánh giá rủi ro, đến chuẩn bị và thực hiện các đề cương để Đánh giá (DQ, IQ, OQ, PQ) ngay từ giai đoạn lập kế hoạch. Điều này giúp giảm bớt gánh nặng cho khách hàng.
Ngoài ra, chúng tôi có thể đáp ứng các yêu cầu về nhiệm vụ Thẩm định riêng, tách biệt với các nhiệm vụ lập kế hoạch và thiết kế. Chúng tôi linh hoạt trong việc phản hồi cho những khách hàng đã hợp tác với chúng tôi trong giai đoạn xây dựng, vì vậy hãy thoải mái tham khảo ý kiến của chúng tôi.
Vui lòng liên hệ với chúng tôi nếu bạn cần tư vấn về vấn đề thẩm định.
Quy trình Thẩm định trong một Dự án
Chuẩn bị Kế hoạch Thẩm định Gốc
Kế hoạch Thẩm định gốc (VMP) là một kế hoạch Thẩm định toàn diện được biên soạn cho một dự án xây dựng nhà máy dược phẩm. Tài liệu này thường ghi lại các mục sau, được điều chỉnh theo nội dung của dự án:
- Chính sách Thẩm định
- Cấu trúc tổ chức cho các hoạt động Thẩm định
- Các hệ thống phải thực hiện Thẩm định (cơ sở, thiết bị, máy móc, v.v.)
- Tổng quan về các quy trình sản xuất
- Định dạng cho các đề cương và báo cáo khác nhau
- Lịch trình Thẩm định
- Kiểm soát thay đổi và quản lý sai lệch
- Tham chiếu đến các tài liệu, v.v.
VMP được tóm tắt trong giai đoạn lập kế hoạch cơ bản của lịch trình tổng thể của dự án, làm rõ chính sách thẩm định cho dự án.

Tiến hành Đánh giá Rủi ro
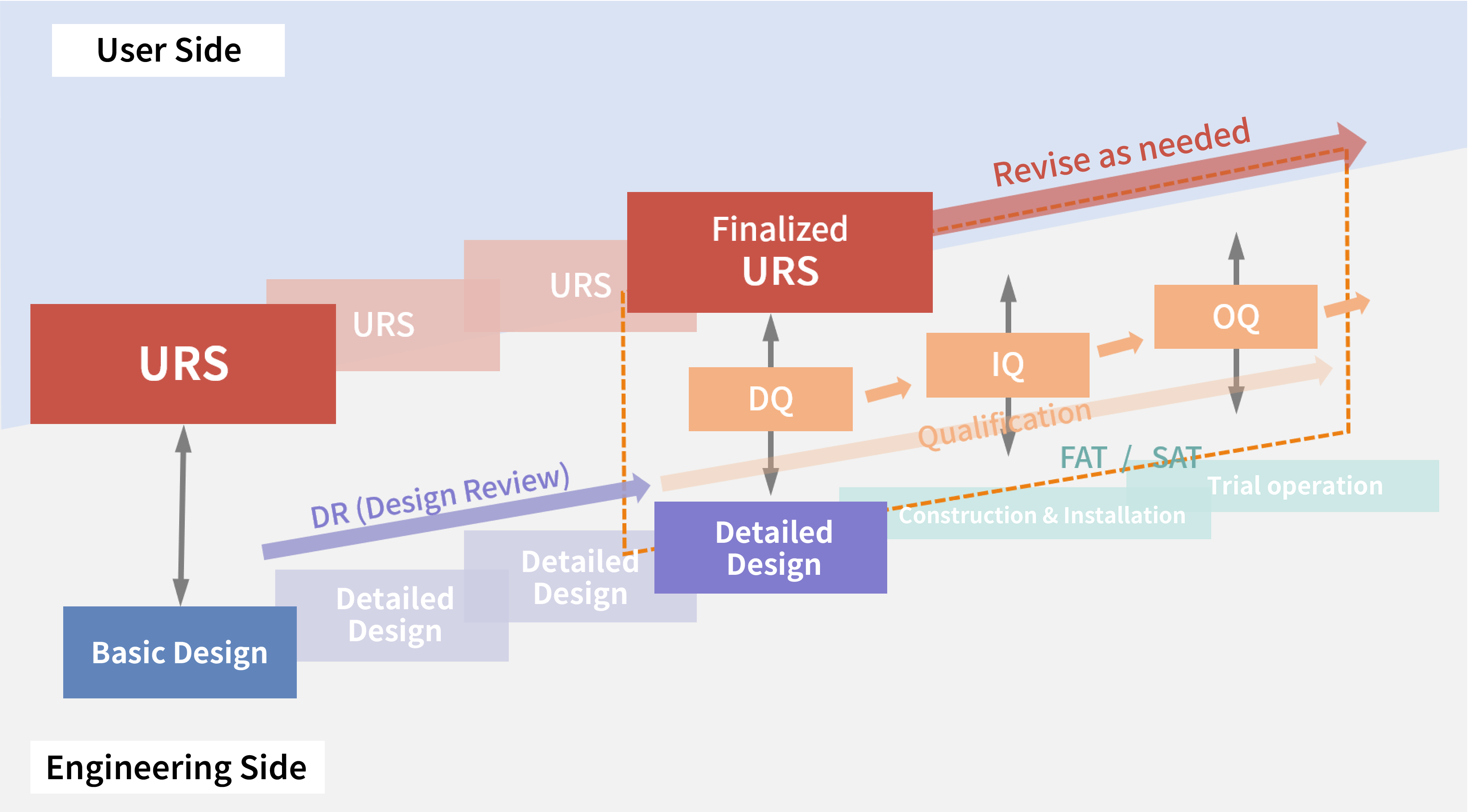
Trong những năm gần đây, phương pháp quản lý rủi ro chất lượng đã được tích hợp vào quy trình lập kế hoạch để thẩm định. Một kế hoạch Thẩm định được tạo ra dựa trên kết quả đánh giá rủi ro. Trong giai đoạn đánh giá rủi ro, phạm vi hệ thống phải để Đánh giá được xác định và nội dung của các mục xác minh sẽ được thực hiện trong DQ, IQ, OQ và PQ được lập kế hoạch.
Thực hiện Đánh giá (DQ, IQ, OQ)
Dựa trên kết quả đánh giá rủi ro, các đề cương theo các kế hoạch Đánh giá khác nhau được tạo ra. Trong giai đoạn thực hiện Đánh giá, điều quan trọng là phải kiểm soát thay đổi và sai lệch một cách hợp lý, đảm bảo các Đánh giá được thực hiện theo các tài liệu thiết kế và đề cương đã được phê duyệt. Gần đây, việc tham chiếu kết quả của các Kiểm tra nghiệm thu tại xưởng (FAT) hoặc Kiểm tra nghiệm thu tại Công trường (SAT) do các nhà cung cấp thực hiện trong IQ và OQ để tiến hành đánh giá đã trở thành thông lệ phổ biến.
Thực hiện PQ, Thẩm định Quy trình, Thẩm định vệ sinh và Thẩm định phương pháp phân tích
Sau khi hoàn thành IOQ, PQ và thẩm định quy trình (PV) cho các sản phẩm được sản xuất sẽ được tiến hành. PQ sử dụng nguyên liệu hoặc placebo được sử dụng trong sản xuất thực tế để xác minh sản xuất trong điều kiện vận hành xấu nhất, lấy mẫu các quá trình quan trọng và trung gian để đảm bảo sản xuất khả thi trong phạm vi vận hành dự kiến. Do có thể sử dụng nguyên liệu đắt tiền cho PQ và PV, nên điều quan trọng là phải sử dụng dữ liệu OQ, xem xét kỹ lưỡng các mục thực hiện PQ và PV để tạo ra các kế hoạchthẩm định hiệu quả và thực hiện chúng một cách hợp lý. Hơn nữa, đối với thẩm định quy trình, thẩm định phương pháp vệ sinh và phương pháp phân tích để đánh giá cũng là điều cần thiết. Sản xuất chỉ có thể bắt đầu khi tất cả các quy trình thẩm định này hoàn tất.
Tại CM Plus…
Xác định phạm vi đánh giá sử dụng phương pháp tiếp cận dựa trên rủi ro

Ở giai đoạn lập kế hoạch thẩm định, trước tiên cần tiến hành đánh giá rủi ro để đánh giá xem các cơ sở và thiết bị (hệ thống) được đưa vào có tác động trực tiếp đến chất lượng sản phẩm và sự an toàn của bệnh nhân hay không. Dựa trên đánh giá này, phạm vi của các hệ thống để đánh giá được xác định (đánh giá hệ thống). Việc tiến hành đánh giá hệ thống chính xác cho phép đánh giá hợp lý, dẫn đến cải thiện chất lượng công việc thẩm định và giảm chi phí không cần thiết.
Soạn thảo Kế hoạch Đánh giá Dựa trên URS
Phụ lục 15 của PIC/S GMP quy định rằng thông số kỹ thuật yêu cầu của người dùng (URS) phải được tham chiếu trong suốt vòng đời thẩm định. Do đó, URS cần được tham chiếu từ các giai đoạn ban đầu của đánh giá . Tại CM Plus, chúng tôi đề xuất soạn thảo kế hoạch đánh giá dự thảo bắt đầu bằng URS. Cách tiếp cận này đảm bảo rằng các yêu cầu được chỉ định trong URS được liên kết trực tiếp với các mục xác minh, làm rõ mối quan hệ giữa các yêu cầu và đánh giá và cho phép tạo ra các kế hoạch đánh giá phù hợp. Hơn nữa, ngay cả trong các hoạt động sau khi triển khai, việc tham chiếu URS cũng tạo điều kiện thuận lợi cho việc quản lý thay đổi phù hợp.
Thẩm định Hệ thống Máy tính /Phần mềm

Chúng tôi cũng cung cấp hỗ trợ để triển khai thẩm định hệ thống máy tính (CSV) và đảm bảo phần mềm máy tính (CSA) cho việc giới thiệu các hệ thống CNTT như Hệ thống Thực hiện Sản xuất (MES) và Hệ thống Quản lý Thông tin Phòng thí nghiệm (LIMS). Phương pháp của chúng tôi tuân thủ ISPE GAMP5 (Phiên bản thứ 2) và hướng dẫn dự thảo CSA của FDA, cung cấp các đề xuất về hoạt động thử nghiệm và tạo kế hoạch dựa trên rủi ro. Chúng tôi cũng cung cấp tư vấn về các yêu cầu về tính toàn vẹn của dữ liệu (bảo mật, theo dõi truy vết, lưu trữ dữ liệu, v.v.) cần thiết cho các hệ thống máy tính.
Quản lý Hồ sơ dựa trên Thực hành Hồ sơ tốt (GDP)

Việc Thẩm định thành công không thể diễn ra nếu không có quản lý tài liệu thành công trong một dự án. Tại CM Plus, chúng tôi cung cấp hướng dẫn và hỗ trợ dựa trên Thực hành tài liệu tốt (GDP), bao gồm quản lý tài liệu của nhà cung cấp và chính thức hóa các quy tắc tạo hồ sơ, để hỗ trợ việc tạo tài liệu tuân thủ GMP. Ngoài ra, chúng tôi hỗ trợ việc tạo tài liệu huấn luyện GMP, đề cương PQ/PV và SOP dựa trên nhu cầu của khách hàng.
-
Để thẩm định nhà máy sản xuất và thiết bị kiến trúc,
pvui lòng tham khảo mục Vận hành & Đánh giá (C&Q) trong dịch vụ kỹ thuật.
Nội dung liên quan


Thông tin hữu ích
Vui lòng tham khảo các bài viết trên trang web phổ biến thông tin của chúng tôi “GMP platform”.
Mặc dù tiêu đề bao gồm “Xây dựng nhà máy dược phẩm”, nhưng nội dung áp dụng cho việc xây dựng trong nhiều ngành công nghiệp khác nhau, không giới hạn ở các nhà máy dược phẩm.

“Bí quyết xây dựng nhà máy dược phẩm”
Kiến thức chuyên môn để hoàn thành thành công các dự án xây dựng nhà máy dược phẩm được biên soạn. Nó bao gồm các hoạt động C&Q/Thẩm định cần được thực hiện ở từng giai đoạn của dự án. Nó giải thích mọi thứ từ việc xây dựng chiến lược C&Q đến DR/DQ, SAT và IQ/OQ.
Chúng tôi đã xuất bản một cuốn sách phiên bản tiếng Anh. Vui lòng tham khảo tin tức này.

“Quản lý dự án của chủ đầu tư trong xây dựng nhà máy dược phẩm, ấn bản lần thứ 2”
Phần thứ 16 trong loạt bài về quản lý dự án xây dựng của chủ sở hữu, tập trung vào trình độ chuyên môn.


