


Validasi dalam Manufaktur Farmasi
Validasi didefinisikan sebagai berikut;
“Program terdokumentasi yang memberikan tingkat kepastian tinggi bahwa suatu proses, metode, atau sistem tertentu akan secara konsisten menghasilkan hasil yang memenuhi kriteria penerimaan yang telah ditentukan sebelumnya. (ICH Q7A)”
“Menetapkan bukti terdokumentasi yang memberikan tingkat kepastian tinggi bahwa suatu proses tertentu akan secara konsisten menghasilkan suatu produk yang memenuhi spesifikasi dan atribut kualitas yang telah ditentukan sebelumnya. (Panduan Validasi Proses FDA 1987)”
Pelaksanaan validasi yang efisien dan berkualitas tinggi merupakan salah satu tantangan terpenting yang harus dicapai dalam proyek pembangunan atau renovasi baru pabrik farmasi. Baru-baru ini, industri farmasi telah beralih dari validasi tradisional ke “validasi berbasis sains dan risiko.” Transisi ini memerlukan penerapan penilaian risiko dan pembuatan rencana validasi berdasarkan penilaian tersebut, yang akan meningkatkan upaya yang diperlukan untuk validasi.
CM Plus menawarkan layanan yang disesuaikan dengan kebutuhan pelanggan, mulai dari dukungan untuk membuat rencana induk validasi dan penilaian risiko, hingga menyiapkan dan melaksanakan protokol untuk kualifikasi (DQ, IQ, OQ, PQ) sejak tahap perencanaan. Hal ini membantu mengurangi beban pelanggan.
Selain itu, kami dapat mengakomodasi permintaan untuk tugas validasi saja, terpisah dari tugas perencanaan dan desain. Kami fleksibel dalam menanggapi pelanggan yang sudah berada dalam tahap konstruksi, jadi jangan ragu untuk berkonsultasi dengan kami.
Jangan ragu untuk menghubungi kami mengenai validasi.
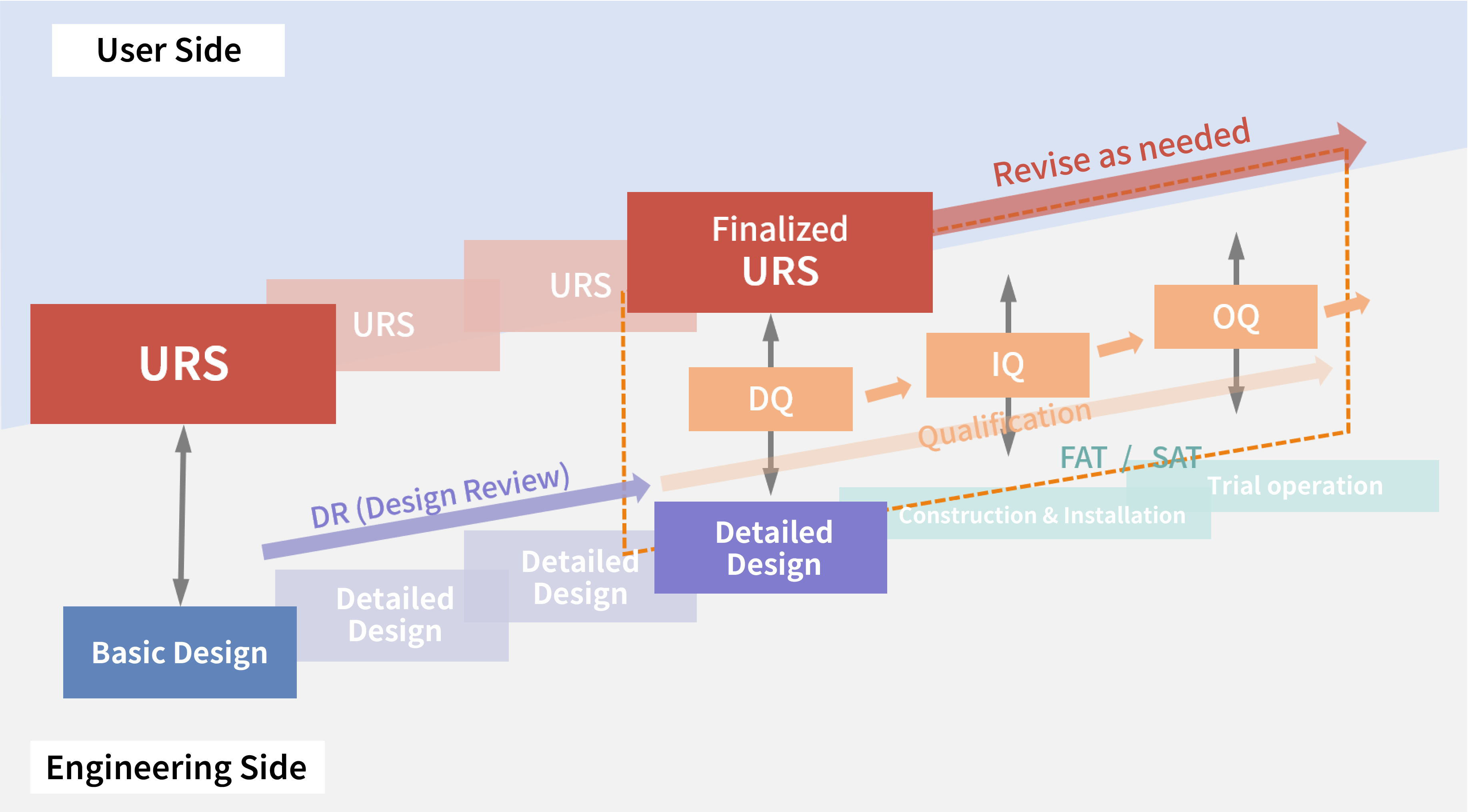
Alur Validasi dalam Proyek
Mempersiapkan Rencana Induk Validasi
Rencana Induk Validasi (VMP) adalah rencana validasi komprehensif yang disusun untuk proyek pembangunan pabrik farmasi. Rencana ini biasanya mendokumentasikan hal-hal berikut, yang disesuaikan dengan isi proyek:
- Kebijakan validasi
- Struktur organisasi untuk aktivitas validasi
- Sistem yang tunduk pada validasi (fasilitas, peralatan, mesin, dll.)
- Tinjauan umum proses manufaktur
- Format untuk berbagai protokol dan laporan
- Jadwal validasi
- Kontrol perubahan dan manajemen deviasi
- Referensi ke dokumen, dll.
VMP dirangkum selama tahap perencanaan dasar dari keseluruhan jadwal proyek, yang mengklarifikasi kebijakan validasi untuk proyek tersebut.

Melakukan Penilaian Risiko
Dalam beberapa tahun terakhir, pendekatan manajemen risiko mutu telah diintegrasikan ke dalam proses perencanaan untuk validasi. Rencana validasi dibuat berdasarkan hasil penilaian risiko. Selama fase penilaian risiko, rentang sistem yang akan dikualifikasi ditentukan, dan konten item verifikasi yang akan dijalankan selama DQ, IQ, OQ, dan PQ direncanakan.
Kualifikasi Pelaksana (DQ, IQ, OQ)
Berdasarkan hasil penilaian risiko, protokol menurut berbagai rencana kualifikasi dibuat. Selama periode pelaksanaan kualifikasi, sangat penting untuk mengelola perubahan dan penyimpangan dengan benar, memastikan kualifikasi dilakukan mengikuti dokumen desain dan protokol yang disetujui. Baru-baru ini, telah menjadi praktik umum untuk merujuk pada hasil Uji Penerimaan Pabrik (FAT) atau Uji Penerimaan Lokasi (SAT) yang dilakukan oleh pemasok dalam kualifikasi IQ dan OQ untuk melakukan kualifikasi.
Melaksanakan PQ, Validasi Proses, Validasi Pembersihan, dan Validasi Metode Analisis
Setelah menyelesaikan IOQ, PQ dan validasi proses (PV) untuk produk yang akan diproduksi dilakukan. PQ menggunakan bahan baku atau plasebo yang digunakan dalam produksi aktual untuk memverifikasi produksi dalam kondisi operasi terburuk, mengambil sampel proses penting dan zat antara untuk memastikan produksi layak dalam rentang operasi yang diharapkan. Mengingat bahan baku yang mahal dapat digunakan untuk PQ dan PV, penting untuk memanfaatkan data OQ, meneliti dengan cermat item pelaksanaan PQ dan PV untuk membuat rencana validasi yang efisien, dan menjalankannya secara rasional. Selain itu, untuk validasi proses, validasi metode pembersihan dan metode analisis untuk evaluasi juga diperlukan. Produksi hanya dapat dimulai setelah semua validasi ini selesai.
Di CM Plus…
Penentuan Cakupan Kualifikasi Menggunakan Pendekatan Berbasis Risiko

Pada tahap perencanaan validasi, penilaian risiko pertama kali dilakukan untuk mengevaluasi apakah fasilitas dan peralatan (sistem) yang akan diperkenalkan berdampak langsung pada kualitas produk dan keselamatan pasien. Berdasarkan evaluasi ini, ruang lingkup sistem yang akan dikualifikasi ditentukan (penilaian sistem). Melakukan penilaian sistem yang akurat memungkinkan kualifikasi yang rasional, yang mengarah pada peningkatan kualitas kerja validasi dan pengurangan biaya yang tidak perlu.
Penyusunan Rencana Kualifikasi Berdasarkan URS
Lampiran 15 dari PIC/S GMP menetapkan bahwa spesifikasi persyaratan pengguna (URS) harus dirujuk sepanjang siklus validasi. Oleh karena itu, URS perlu dirujuk sejak tahap awal kualifikasi. Di CM Plus, kami mengusulkan penyusunan rencana kualifikasi dimulai dengan URS. Pendekatan ini memastikan bahwa persyaratan yang ditetapkan dalam URS secara langsung terkait dengan item verifikasi, sehingga hubungan antara persyaratan dan kualifikasi menjadi jelas dan memungkinkan pembuatan rencana kualifikasi yang sesuai. Lebih jauh, bahkan selama operasi pasca-implementasi, merujuk ke URS memfasilitasi manajemen perubahan yang tepat.
Validasi untuk Sistem Komputer/Perangkat Lunak

Kami juga menyediakan dukungan untuk penerapan validasi sistem terkomputerisasi (CSV) dan jaminan perangkat lunak komputer (CSA) untuk pengenalan sistem TI seperti Sistem Eksekusi Manufaktur (MES) dan Sistem Manajemen Informasi Laboratorium (LIMS). Pendekatan kami sesuai dengan ISPE GAMP5 (Edisi ke-2) dan rancangan panduan CSA FDA, yang menyediakan pembuatan rencana berbasis risiko dan proposal aktivitas pengujian. Kami juga menawarkan saran tentang persyaratan integritas data (keamanan, jejak audit, penyimpanan data, dll.) yang diperlukan untuk sistem komputer.
Manajemen Dokumen Berdasarkan Good Document Practice (GDP)

Validasi yang berhasil tidak dapat terjadi tanpa manajemen dokumen yang berhasil dalam suatu proyek. Di CM Plus, kami menyediakan panduan dan dukungan berdasarkan Praktik Dokumen yang Baik (GDP), termasuk manajemen dokumen pemasok dan formalisasi aturan pembuatan catatan, untuk mendukung pembuatan dokumen yang sesuai dengan GMP. Selain itu, kami mendukung pembuatan materi edukasi GMP, protokol PQ/PV, dan SOP berdasarkan kebutuhan pelanggan.
-
Untuk validasi fasilitas manufaktur dan peralatan arsitektur,
silakan merujuk ke Commissioning & Qualification (C&Q) dalam layanan teknik.
Konten Terkait


Informasi Berguna
Silakan merujuk pada artikel di situs penyebaran informasi kami “Platform GMP.”
Meskipun judulnya mencakup “Konstruksi Pabrik Farmasi,” kontennya berlaku untuk konstruksi di berbagai industri, tidak terbatas pada pabrik farmasi.

“Cara Membangun Pabrik Farmasi”
Pengetahuan untuk menyelesaikan proyek pembangunan pabrik farmasi dengan sukses dihimpun. Pengetahuan ini mencakup aktivitas C&Q/Validasi yang harus dilakukan pada setiap tahap proyek. Pengetahuan ini menjelaskan semuanya mulai dari formulasi strategi C&Q hingga DR/DQ, SAT, dan IQ/OQ.
Kami telah menerbitkan buku dalam bahasa Inggris. Silakan lihat berita ini.

“Manajemen Proyek Pemilik dalam Konstruksi Pabrik Farmasi Edisi ke-2”
Angsuran ke-16 dalam seri tentang manajemen proyek konstruksi oleh pemilik, dengan fokus pada kualifikasi.


